Abstract:
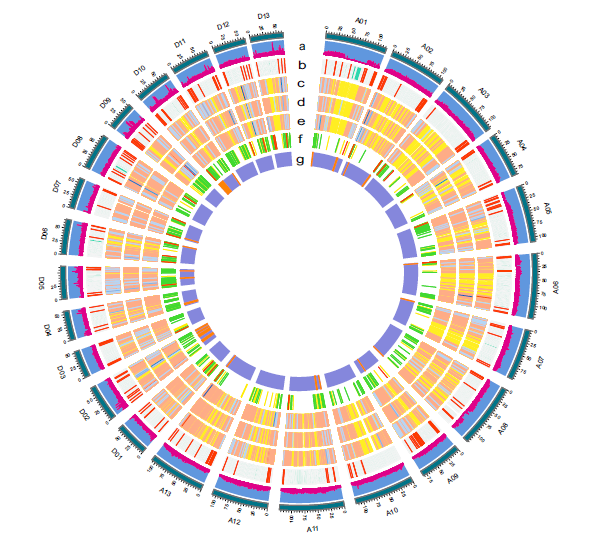
Interspecific breeding in cotton takes advantage of genetic recombination among desirable genes from different parental lines. However, the expression new alleles (ENAs) from crossovers within genic regions and their significance in fibre length (FL) improvement are currently notunderstood. Here, we generated resequencing genomes of 191 interspecific backcross inbredlines derived from CRI36 (Gossypium hirsutum)9Hai7124 (Gossypium barbadense) and 277dynamic fibre transcriptomes to identify the ENAs and extremely expressed genes (eGenes)potentially influencing FL, and uncovered the dynamic regulatory network of fibre elongation. Of35 420 eGenes in developing fibres, 10 366 ENAs were identified and preferentially distributedin chromosomes subtelomeric regions. In total, 1056–1255 ENAs showed transgressiveexpression in fibres at 5–15 dpa (days post-anthesis) of some BILs, 520 of which were located inFL-quantitative trait locus (QTLs) andGhFLA9(recombination allele) was identified with a largereffect for FL thanGhFLA9of CRI36 allele. Using ENAs as a type of markers, we identified threenovel FL-QTLs. Additionally, 456 extremely eGenes were identified that were preferentiallydistributed in recombination hotspots. Importantly, 34 of them were significantly associated withFL. Gene expression quantitative trait locus analysis identified 1286, 1089 and 1059 eGenes thatwere colocalized with the FL trait at 5, 10 and 15 dpa, respectively. Finally, we verified theGhir_D10G011050gene linked to fibre elongation by the CRISPR-cas9 system. This studyprovides the first glimpse into the occurrence, distribution and expression of the developingfibres genes (especially ENAs) in an introgression population, and their possible biological significance in FL
Keywords:Gossypium, Intragenicrecombination, Fibre length,Expression new alleles, Extremelyexpressed gen